When we delve into the world of atoms and molecules, we encounter a complex quantum system where positively charged nuclei are surrounded by negatively charged electrons. The interactions between these electrons of multiple atoms when they come together to form a molecule are incredibly intricate. This complexity has made the computer simulation of molecules one of the most challenging problems in modern science.
Recently, researchers from the Berlin Institute for the Foundations of Learning and Data (BIFOLD) at TU Berlin and Google DeepMind have made a significant breakthrough in this field. They have developed a new machine learning algorithm that allows for highly accurate simulations of single or multiple molecules over long time-scales. This groundbreaking work has been published in Nature Communications.
The Importance of Molecular Dynamics Simulations
Molecular dynamics simulations play a crucial role in understanding the properties of molecules and materials. Furthermore, these simulations have vast applications in various fields such as drug development and material design for technologies like solar panels and batteries. Traditionally, computing the interactions of electrons in molecules involved solving the Schrödinger equation. However, this task is incredibly challenging, especially for molecules with more than a few dozen atoms.
Overcoming Computational Challenges with Machine Learning
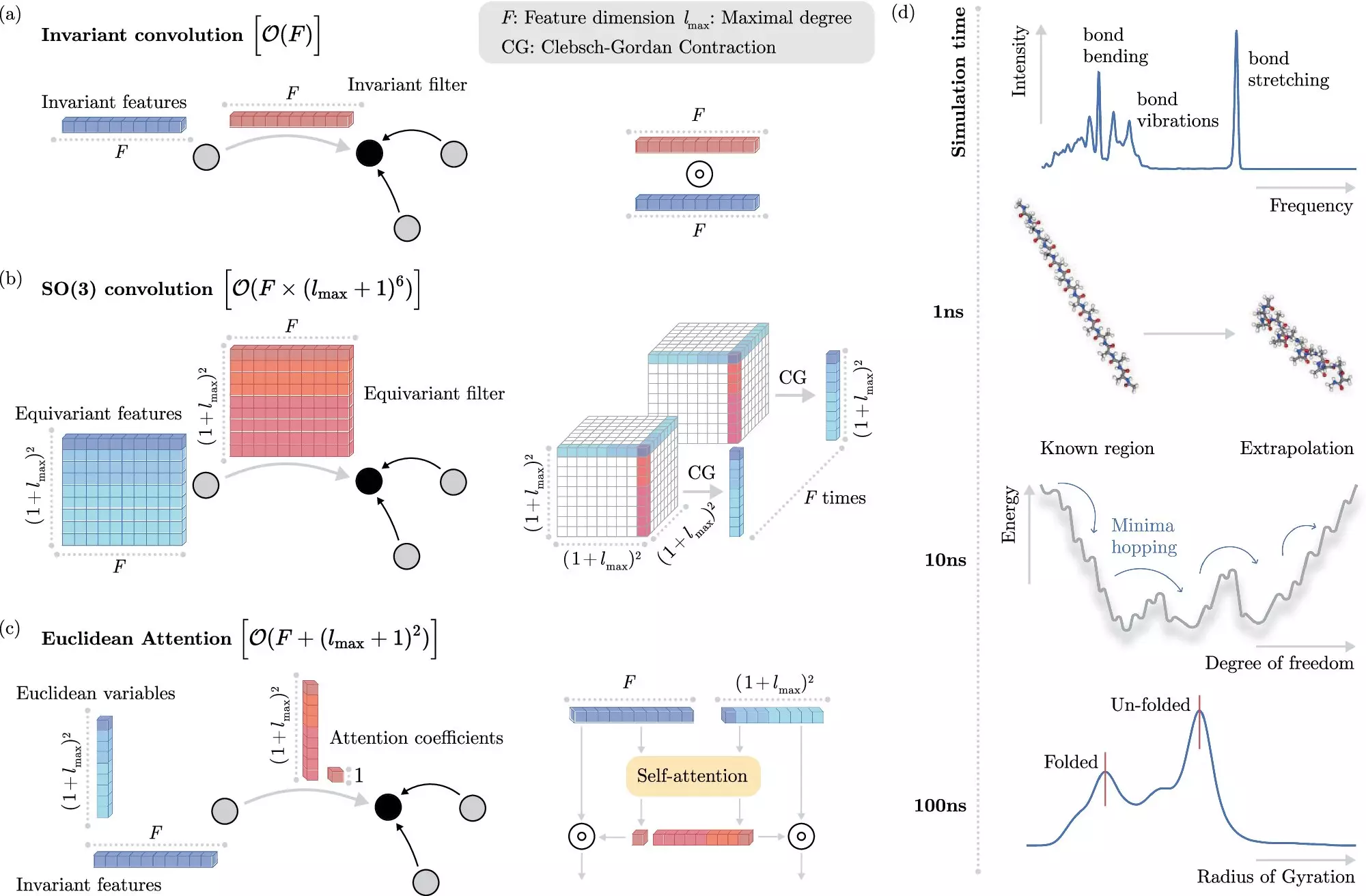
Machine learning methods have revolutionized this field by providing an alternative approach to solving the Schrödinger equation. Instead of explicitly solving the equation, these methods can predict electronic interactions at the atomistic level with significantly reduced computational cost. The key challenge lies in efficiently teaching the machine learning system how electrons interact without explicit modeling.
Improving Efficiency in Molecular Dynamics Simulations
The BIFOLD scientists have introduced a new learning algorithm that decouples invariances from other information about a chemical system. By simplifying this process, the model can focus on the most critical operations, reducing the overall computational cost significantly. This advancement allows for simulations that previously required months or years of computation to be completed within days on a single computer node.
This breakthrough opens up a world of possibilities in the field of molecular dynamics simulations. Researchers can now accurately simulate interactions between molecules and proteins in the human body, potentially leading to the development of new drugs without the need for extensive experiments. By combining advanced machine learning techniques with physical principles, researchers can overcome long-standing challenges in computational chemistry.
The fusion of machine learning with the study of atoms and molecules represents a significant leap forward in our understanding of the fundamental processes of nature. With the ability to simulate complex systems more accurately and efficiently, researchers are poised to unlock new frontiers in drug development, material design, and various other scientific endeavors. As we look towards the future, the potential for further breakthroughs in molecular dynamics simulations is limitless.
Leave a Reply